
European doctors and their patients have frequently had access to new technologies before their American counterparts. In large part, this has been due to what was perceived as a saner and simpler regulatory process in the European Union than in the United States. But the EU regulatory process is changing, and that may have long-term repercussions for European surgeons, patients, and investors.
Ignorance of regulatory pathways can scuttle the best-made plans for bringing innovative technologies to market. Technical excellence alone is insufficient to meet the challenge of launching a medical device; companies also need first-class regulatory and quality-control expertise. Although that is true around the globe, it has often been said that the regulatory process is more rational in the European Union than in the United States. But is this true?
Companies have generally found the process of launching new medical devices in the United States more challenging than in the European Union. However, that may be changing because of tightened EU regulations for the approval and certification of medical devices with the EU's new Medical Devices Directive (MDD).
CHANGING LANDSCAPE
Historically, medical device companies have criticized the FDA for stifling innovation through inconsistent regulation and a slow approval process. Challenges in the US regulatory environment have sometimes prompted investors to shift their funds overseas—often to Europe, where the regulatory process has been less bureaucratic, more efficient, and more predictable.
The FDA has required device makers to provide evidence of both the safety and efficacy of a device, whereas the CE Mark has required only proof of safety and proof that the device performs in a manner consistent with the manufacturer's intended use.
Despite any criticisms, the FDA process is not without an upside for industry. Once a device has received FDA clearance, companies can start marketing their product across the entire country. Furthermore, reimbursement in the United States, such as through Medicare, is more consistent than in the European Union, where each member state may require its own form of proof of efficacy before granting reimbursement. Even with a CE Mark, companies have no guarantee that use of a given device will be reimbursable in any given country.
The Road Ahead

1. There are significant inconsistencies in policies among the three directorates-generals that oversee the device approval process: the directorates of Communications Networks, Content, and Technology; of Health and Consumers; and of Justice. These inconsistencies are leading to conflicting policies that have a negative impact on innovation.
2. Leaving the classification of each medical device to each member state impedes the goal of EU harmonization.
3. Recent discussions on whether software applications should be considered parts of medical devices, and regulated as such, further breaks apart what should ideally be a unified decision-making process for notified bodies.
4. Although the European Union continues to make innovation-friendly statements, inconsistencies in current policy make the job of navigating the EU regulatory process an impediment to medical device innovation.
THE FDA ROUTE
In order to have a medical device approved in the United States, the device must first be classified. Class 1 includes low-risk products such as bandages and sunglasses. Class 2 includes products involving medium risk, such as bone-fixing screws. Class 3 includes high-risk products, such as pacemakers (Table 1).
Information on the FDA website (www.fda.gov/medicaldevices) can help companies determine how a new product is likely to be classified. After identifying similar products listed on the site, a company can determine whether its product is likely to fall into an exemption category that will allow use of a 510(k) approval process (ie, showing equivalence to a product or products already on the market) or whether the product will require full-blown premarket approval (PMA).
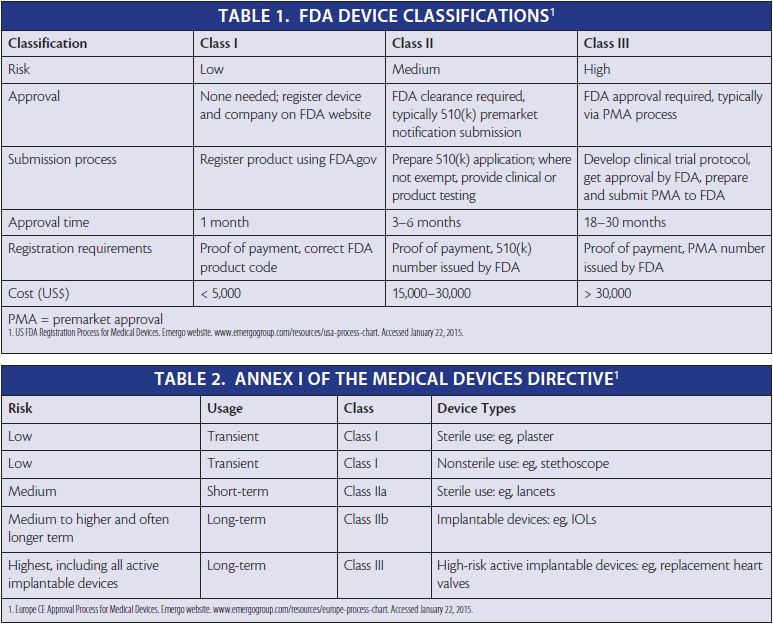
The US regulatory process depends heavily on whether predicate devices have already been registered in the United States. A predicate device is a legally marketed device deemed substantially equivalent to the device under investigation. Substantial equivalence is determined on the basis of intended use, design, energy used or delivered, materials, performance, safety, effectiveness, labeling, biocompatibility, standards, and other applicable measures. Where no predicate device exists, companies must use code section 513(g) for a de novo process, which causes costs to escalate rapidly in terms of both time and money.
THE CE ROUTE
In the past few years, two issues—device quality and patient risk—have taken center stage in the EU device approval process. Patient safety became a public concern following several high-profile failures in the medical device industry, including the Poly Implant Prothèse (PIP) breast implant scandal of 2010, in which the manufacturer substituted industrial grade silicone oil for medical grade.1 Considerable harm was done to patients when some of the implants ruptured—and harm was done as well to the public's perception of the EU regulatory process for medical devices.
Now, companies that neglect issues of quality and safety during the early stages of product development in the European Union may pay a heavy price as they reach the later approval stages, when they come into contact with regulatory authorities.
Bringing medical devices to the EU market involves compliance with the basic requirements outlined in the EU's MDD or Active Implantable Medical Devices Directive (AIMDD), as well as adherence to harmonized standards such as those detailed in quality systems norms.
Although the FDA route has often been criticized for bureaucratic inefficiency, the EU approval process is also bureaucratically complex and involves multiple stakeholders, including the manufacturer of the device and its subcontractors and distributors, authorized representatives, competent authorities, and notified bodies (see Glossary of Key CE Mark Stakeholders).
The first step in developing a medical device for release in the EU is to determine its classification according to Annex I of the MDD, which is based on the intended use, risk of the device class, duration of contact with the patient, degree of invasiveness, and part of the body contacted (Table 2). Unfortunately, this must be done on a country-by-country basis. There is no guarantee, for example, that France will classify a device the same way as Denmark; the classification depends on the view of the national competent authority.
AT A GLANCE
• Whether in the United States or the European Union, bringing ophthalmic products to market requires expertise to navigate challenging regulatory pathways. • Increasingly, issues of device quality and patient risk have become important in EU medical device regulation.
• Companies that neglect issues of quality control and risk reduction during the early stages of product development in the European Union may pay a price as they reach the later approval stages, when they first come into contact with regulatory authorities.
• Changes in EU regulatory directives and the slow legislative process are confusing the regulatory pathway and making the release of innovative products a growing challenge.
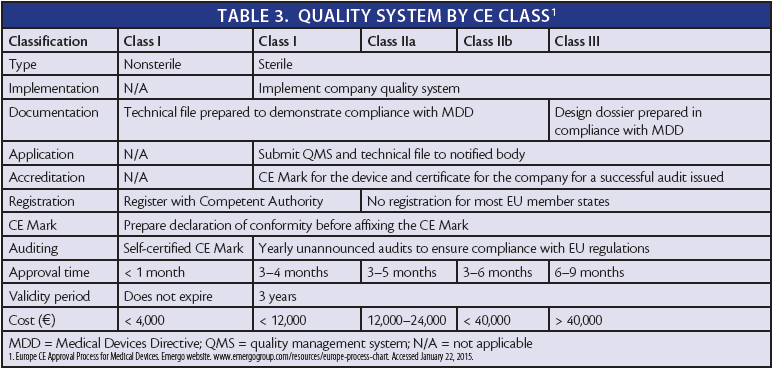
Once the classification has been established in a given member state, the manufacturer must implement a quality system and prepare a technical file and design dossier (Table 3). The notified body will then audit the manufacturer's quality system and technical file. After the device passes the audit, the manufacturer must register the device with the competent authority and prepare the declaration of conformity so that the CE Mark can be affixed to the device.
As the product is developed, the company must show evidence of a continuously updated quality system and application of updates to the quality manual as required. This can be demonstrated through the assignment and training of staff and through conducting periodic gap analyses of the quality system through internal audits. Documentation of preventive actions helps to demonstrate that this continuous improvement process is being undertaken.
Glossary of Key CE Mark Stakeholders1
Competent Authorities Charged by the EU member states to enforce the MDD at a national level. Each member state will have its own interpretation of the directives, defining how the competent authorities should implement the MDD. Competent authorities are responsible for market surveillance, field safety corrective actions (recalls), and accrediting notified bodies.
Notified Bodies Audit manufacturers' quality systems, test devices for compliance to applicable directives and standards, and assess the conformity of the submitted technical files and design dossiers before a CE certification is approved.
Authorized Representatives Required for any company without a physical presence in the European Union. Authorized representatives act as points of contact between manufacturers and competent authorities.
- Council Directive 93/42/EEC CONSLEG 1993L0042. European Union website. http://eur-lex.europa.eu/ LexUriServ/LexUriServ.do?uri=CONSLEG:1993L0042:20071011:en:PDF. Accessed January 22, 2015.
CONCLUSION
A combination of changes in EU regulatory directives and a stagnant European legislative process related to medical devices is making the release of innovative products a growing challenge in the European Union. Several evident problems are outlined in The Road Ahead on the opening page of this article. A continuation of these problems will threaten the European Union's global competitive edge in technological development. n
1. Pips breast implant scandal: Regulator warned years earlier. May 15, 2012. The Telegraph. http://www.telegraph.co.uk/health/...../Pips-breast-implant-scandal-Regulator-warned-years-earlier.html. Accessed January 5, 2015.
Yijun Huang, PhD
- Cofounder and Chief Technology Officer, EyeKor, Madison, Wisconsin
- yhuang@eyekor.com
- Financial disclosure: Owner, Shareholder (EyeKor)
Mark S. Talary, PhD
Chief Technology Officer, IROC Science, Zurich, Switzerland
- mark.talary@irocscience.com
- Financial disclosure: Salaried by company (IROC Science)