



Now and again, a patient may have a unique, specific, and serious need that is unmet by currently available FDA-approved options. When a viable option is available outside the United States, the FDA has a pathway to expanded access that is sometimes referred to as the compassionate use device exemption (CUDE). The pathway is reserved for patients with an “immediately life-threatening condition or serious disease or condition to gain access to an investigational medical product (drug, biologic, or medical device) for treatment outside of clinical trials when no comparable or satisfactory alternative therapy options are available.”1 This is a high threshold to meet, as it should be. CUDE is designed for special cases, not as an end run around normal approval processes.
According to the FDA, “serious disease or condition means a disease or condition associated with morbidity that has substantial impact on day-to-day functioning. Short-lived and self-limiting morbidity will usually not be sufficient, but the morbidity need not be irreversible, provided it is persistent or recurrent. Whether a disease or condition is serious is a matter of clinical judgment, based on its impact on such factors as survival, day-to-day functioning, or the likelihood that the disease, if left untreated, will progress from a less severe condition to a more serious one.”2
Rarely, an ophthalmic case warrants expanded access. Each CUDE is for a single person only and is not intended as a substitute for an investigational device exemption, which should be pursued if a large need is identified. This article shares a few representative cases from the Cincinnati Eye Institute in which a CUDE was the patient’s viable option.
CASE NO. 1
After undergoing cataract surgery and the implantation of a 4.00 D hydrophilic acrylic IOL, the patient experienced disabling halos, glare, and reflections. The resting entrance pupil size was 7 mm under ambient illumination, equating to a true pupil size at the iris plane of roughly 6.25 mm and exposing the IOL edges to ambient light, especially in low light. The instillation of pupil-modulating drops caused intolerably dim vision. The high reflectivity of the hydrophobic acrylic IOL in situ compounded the patient’s symptoms. The fellow eye had intolerable and disabling glare from a posterior subcapsular cataract.
Expanded access to an oversized, low-powered IOL with a lower index of refraction than that of the IOL in the patient’s eye from a European manufacturer was sought and was granted by the FDA. The product manufacturer provided concurrence, and an independent Institutional Review Board agreed to monitor the case.
The posterior subcapsular cataract was extracted, and an oversized IOL was implanted. The patient achieved a favorable result. Postoperatively, he asked, “Can I give you a hug?”
The IOL in the first eye was exchanged for the same oversized implant, and a similarly successful result was obtained.
CASE NO. 2
A patient with ocular melanoma underwent radiation therapy. A mature cataract gradually formed over the next few years. The patient underwent cataract surgery and was found afterward to have radiation retinopathy. Vision in the operative eye was markedly reduced such that she could perceive only shapes and shadows. The patient experienced disabling visual confusion between her two eyes and binocular diplopia that presented as shadow images. She was intolerant to wearing an opaque contact lens on the affected eye and was adamantly opposed to enucleation.
A CUDE for an opaque, black PMMA sulcus-fixated IOL was requested and approved. A unique feature of the device is that the opaque, black PMMA is transparent to infrared light, which makes monitoring of the posterior segment with infrared fundus photography possible using OCT imaging, an indocyanine green camera system, or the like. After receiving the IOL, the patient’s symptoms resolved completely (Figure 1). The opaque implant used in this case is no longer available, owing to market pressures on niche devices.
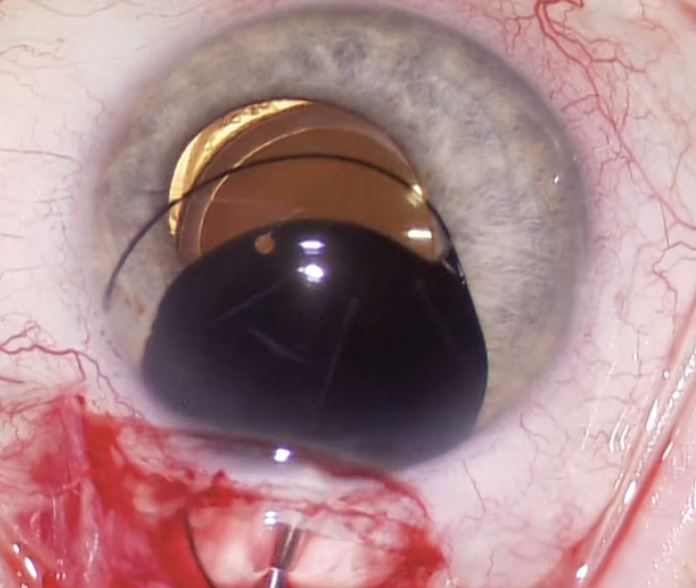
Figure 1. An opaque black IOL is placed in the ciliary sulcus to alleviate binocular shadow diplopia.
CASE NO. 3
In 2007, a 3½-year-old girl presented with severe photophobia. The patient had been born with a congenital cataract and had a history of an optical iridectomy. Light sensitivity caused her to close the affected eye whenever she was outside.
The patient underwent cataract surgery and IOL implantation. Postoperatively, the forced eyelid closure persisted, and amblyopia therapy became progressively less effective. A pupillary cerclage was performed, and the photic symptoms were alleviated. The patient began to make progress with amblyopia therapy.
Approximately 1.5 years after the cerclage procedure, the patient was again observed to be experiencing photophobia and forced eyelid closure outdoors. A cheese-wiring effect with the cerclage suture was noted on examination. Iris repair with sutures was performed but achieved only transient success.
A CUDE for a custom, flexible iris prosthesis was requested and granted. After placement of the device, the patient experienced a complete and sustained resolution of symptoms (Figure 2).
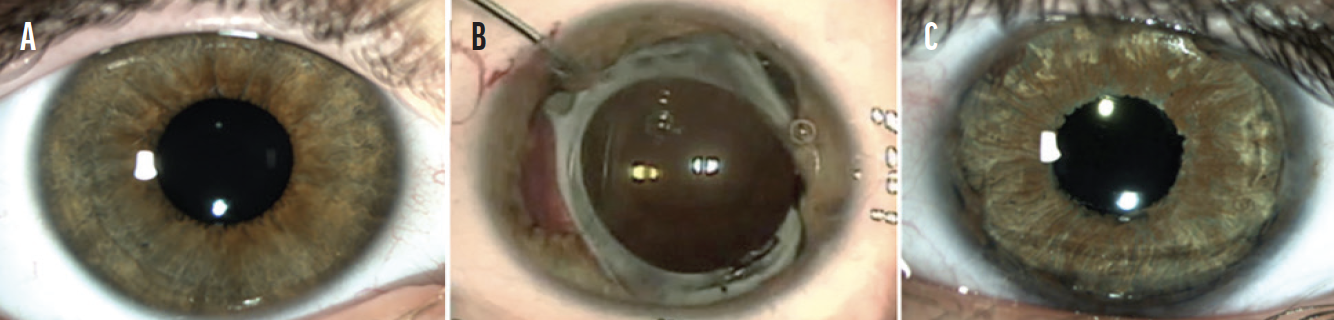
Figure 2. The unaffected eye (A). The appearance of the affected eye after a second failed iris repair (B) and after the placement of a customized, flexible iris prosthesis (C).
A few years after the case, the custom, flexible iris implant device became the subject of a formal investigational device exemption, and it received FDA approval in 2018. The device is now commercially available in the United States as the CustomFlex ArtificialIris (VEO Ophthalmics).
CONCLUSION
CUDEs are rarely necessary. US ophthalmologists are fortunate, however, that the pathway exists for special patients when the need arises. Using the pathway entails the completion of regulatory documentation, reporting obligations, and coordination with disparate entities such as the FDA, the manufacturer, an independent Institutional Review Board, and the surgical facility at which the procedure may be performed. The time-consuming process can be highly rewarding for the patient and the surgeon.
1. Expanded access. US Food & Drug Administration. March 23, 2021. Accessed January 25, 2022. https://www.fda.gov/news-events/public-health-focus/expanded-access
2. CFR – Code of Federal Regulations Title 21. US Food & Drug Administration. October 1, 2021. Accessed January 25, 2022. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=312.300
