



Corneal dystrophies are characterized as bilateral, progressive opacities or changes within the cornea typically following a dominant pattern of inheritance. As per the second edition of the IC3D classification, pre-Descemet corneal dystrophies (PDCDs) fall into category 4—emerging, novel, or previously identified corneal dystrophies for which evidence supporting a distinct entity remains unconvincing. PDCDs comprise distinct subgroups that are potentially sporadic, age-related, or degenerative in nature. Genetic studies provide a means of distinguishing between dystrophic and degenerative disorders. A particular subtype of PDCD is dominant pre-Descemet polychromatic punctate dystrophy (PPDCD).
PPDCD manifests as punctate, polychromatic, pre-Descemet, fine deposits. They can be visualized via direct and indirect illumination. The opacities are uniformly distributed throughout the cornea, from limbus to limbus, with clear spaces between them. PPDCD is inherited in an autosomal dominant manner. Most patients remain asymptomatic. Despite the disease’s progressive nature, patients generally retain vision, and their overall visual prognosis remains favorable.1,2
Potential differential diagnoses include cornea farinata, deep filiform dystrophy, deep punctate dystrophy, Schnyder corneal dystrophy, cystinosis, Bietti dystrophy, and monoclonal gammopathy.3,4
The following case report centers on a PDCD first described by Fernandez Sasso, MD, and colleagues in 1979.5 They described a 36-year-old woman with mild myopia who presented for a routine ophthalmologic examination. Remarkably, seven of her family members were identified with the same corneal dystrophy.
CASE REPORT
A 25-year-old woman presented for an ophthalmologic evaluation. She had a general medical history of hypercholesterolemia and was on a regimen of oral statin therapy. Her visual acuity was 20/20 OU, and her IOP was 14 mm Hg OU.
A slit-lamp examination found multiple symmetrical, pre-Descemet polychromatic punctate deposits bilaterally (Figure 1). A fundoscopic examination revealed no significant alterations in the posterior segment of either eye. For a more detailed view of the corneal architecture, anterior segment OCT (AS-OCT) imaging and confocal microscopy were performed.
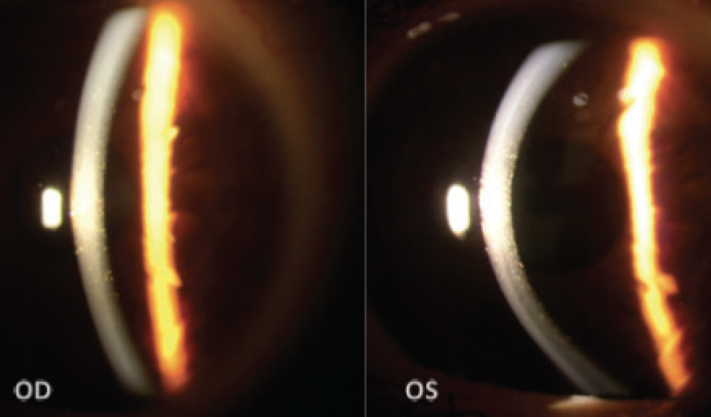
Figure 1. Slit-lamp images show multiple pre-Descemet polychromatic punctate deposits that are symmetrically and evenly distributed in both eyes.
The patient disclosed a familial predisposition to corneal abnormalities. At least four members of her immediate family had similar conditions.
CLINICAL FINDINGS
AS-OCT imaging. Slight hyperreflective dotting was uniformly distributed across the deep pre-Descemet stroma (Figure 2).
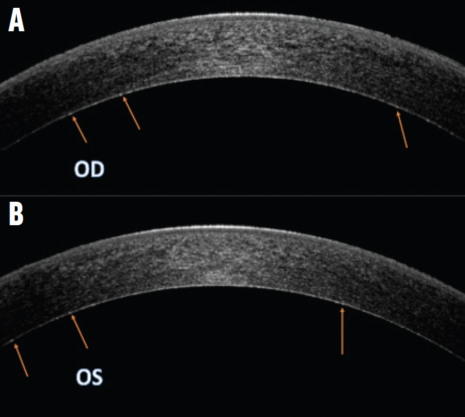
Figure 2. OCT images of the right (A) and left (B) eyes demonstrate a faint, uniformly distributed, hyperreflective dotting in the pre-Descemet stroma. Arrows highlight the deposits in the pre-Descemet region.
Confocal microscopy. No alterations were observed at the level of the epithelium, subbasal nerve plexus, or middle stroma (nerve). In the deep pre-Descemet stroma, however, hyperreflective deposits were noted. These were of a homogeneous shape and size and had blunt edges, as is typically associated with lipid crystals (Figure 3, at outset of article).
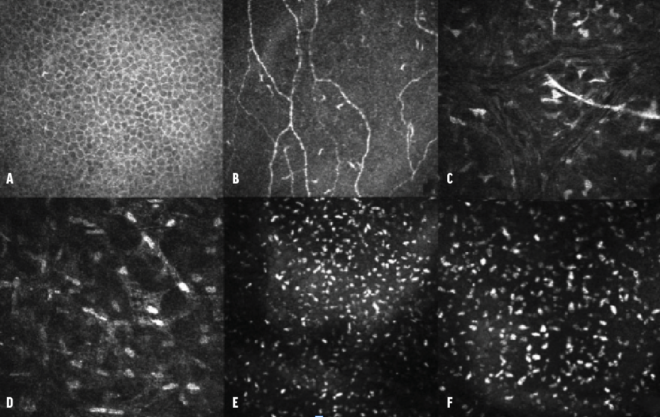
Figure 3. Confocal microscopy series of the epithelium (A), subbasal nerve plexus (B), middle stroma (C), deep stroma (D), and pre-Descemet stroma (E). The images capture multiple hyperreflective deposits located behind the hyperreflectivity of the endothelium. An image obtained with a 300x field objective showcases these deposits in greater detail (F).
CONCLUSION
PPDCD is a rare and distinct disorder distinguished by a well-defined clinical presentation. This sets it apart from other pre-Descemet dystrophies and underscores the importance of careful differential diagnosis.
Of note, patients with PPDCD are generally asymptomatic, maintain good visual acuity, and can expect an excellent prognosis. The typical clinical approach to these cases is observation and follow-up.
Two noninvasive diagnostic tools that have proven instrumental in studying PPDCD are AS-OCT and high-resolution confocal microscopy. In AS-OCT studies, faint hyperreflective opacities have been observed. High-resolution confocal microscopy, on the other hand, helps identify the location and morphology of deposits at a cellular level.6
Another essential aspect of managing PPDCD is genetic counseling. The disease has been associated with a new mutation variant in the PRDX3 gene. Further screening should clarify whether this PRDX3 variant is consistent across all affected families or if locus heterogeneity exists for PPDCD.
A reclassification of PPDCD in the IC3D to a category 1 dystrophy is under consideration. Category 1 dystrophy is characterized as a well-defined corneal dystrophy for which the gene has been mapped and identified and the specific mutations are known. The potential reclassification is due to the distinct clinical presentation of PPDCD, its known association with a particular gene, and its autosomal dominant inheritance pattern.7
1. Henríquez-Recine MA, Marquina-Lima KS, Vallespín-García E, et al. Heredity and in vivo confocal microscopy of punctiform and polychromatic pre-Descemet dystrophy. Graefes Arch Clin Exp Ophthalmol. 2018;256(9):1661-1667.
2. Dolz-Marco R, Gallego-Pinazo R, Pinazo-Durán MD, Díaz-Llopis M. Crystalline subtype of pre-Descemetic corneal dystrophy. J Ophthalmic Vis Res. 2014;9(2):269-271.
3. Lagrou L, Midgley J, Romanchuk KG. Punctiform and polychromatophilic dominant pre-Descemet corneal dystrophy. Cornea. 2016;35(4):572-575.
4. Benito-Pascual B, Arriola-Villalobos P, Díaz-Valle D, Del Castillo-Sánchez JM. Confocal biomicroscopy in four patients with polychromatic corneal dystrophy. Arch Soc Esp Oftalmol. 2018;93(10):470-475.
5. Fernandez-Sasso D, Acosta JE, Malbran ES. Punctiform and polychromatic pre-Descemet’s dominant corneal dystrophy. Br J Ophthalmol. 1979;63(5):336-338.
6. Alafaleq M, Georgeon C, Grieve K, Borderie VM. Multimodal imaging of pre-Descemet corneal dystrophy. Eur J Ophthalmol. 2020;30(5):908-916.
7. Alió del Barrio JL, Chung DD, Al-Shymali O, et al. Punctiform and polychromatic pre-Descemet corneal dystrophy: clinical evaluation and identification of the genetic basis. Am J Ophthalmol. 2020;212:88-97.



